Boston Scientific accounts are for healthcare professionals only.

Nalu Medical is now part of Boston Scientific
Browse product categories

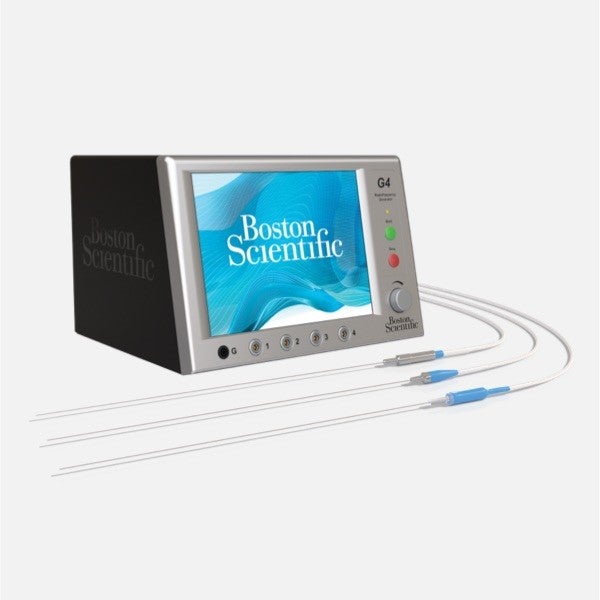
Why choose Boston Scientific as your pain management partner
Unlock more targeted treatment options for your chronic pain patients
We are advancing pain management by providing an essential selection of advanced therapies to treat chronic pain. Explore our latest innovations to help expand access to care and treat more patients.
FBSS, DPN, NSBP, CRPS or Radiculopathy
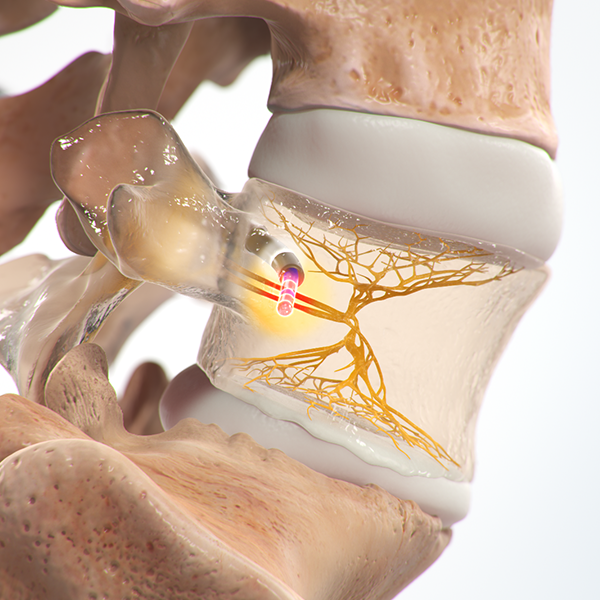
Vertebrogenic Pain
Facetogenic/Joint pain
MRI Conditionality
Boston Scientific’s spinal cord stimulator systems with ImageReady™ MRI conditionality allow patients to safely undergo MRI scans.
Pain management backed by extensive clinical evidence
Multiple Level 1 RCTs and Real-World studies support the design, efficacy, and efficiency of Boston Scientific's pain management therapies.1-6
To learn more about Intracept Procedure training request more information
Tools and resources

Reimbursement
Get reimbursement tools for physicians and administrators.

Education for your patients
Pain.com offers personalized content and connection to real people who can answer questions about pain management therapies.
**Other RFA applications include Neck, SI Joint, Hip, Knee, Foot and more
References:
1. Wallace MS, North JM, Phillips GM, et al. Combination therapy with simultaneous delivery of spinal cord stimulation modalities: COMBO randomized controlled trial. Pain Manag. 2023;13(3):171-184. (N=89)
2. Thomson SJ, Tavakkolizadeh M, Love-Jones S, et al. Effects of rate on analgesia in kilohertz frequency spinal cord stimulation: results of the proco randomized controlled trial. Neuromodulation. 2018;21(1):67-76.
3. North J, Loudermilk E, Lee A, et al. Outcomes of a multicenter, prospective, crossover, randomized controlled trial evaluating subperception spinal cord stimulation at ≤1. 2 khz in previously implanted subjects. Neuromodulation. 2020;23(1):102-108.
4. Metzger CS, Hammond MB, Pyles ST, et al. Pain relief outcomes using an SCS device capable of delivering combination therapy with advanced waveforms and field shapes. Expert Rev Med Devices. 2020;17(9):951-957.
5. Paz-Solís J, Thomson S, Jain R, Chen L, Huertas I, Doan Q. Exploration of high- and low-frequency options for subperception spinal cord stimulation using neural dosing parameter relationships: the halo study. Neuromodulation. 2022;25(1):94-102.
6. Veizi E, Hayek SM, North J, et al. Spinal cord stimulation (Scs) with anatomically guided (3d) neural targeting shows superior chronic axial low back pain relief compared to traditional scs-lumina study. Pain Med. 2017;18(8):1534-1548.
7. Provenzano D, et al. Significant Pain Relief and Treatment Satisfaction Following Radiofrequency Ablation Prospective, Multicenter Study (RAPID) [Abstract]. Fifth Annual Meeting of the American Society of Pain and Neuroscience Society, July 13-16, 2023 (N=289 as of July 2023, ongoing study)
8. Fischgrund J, Rhyne A, Macadaeg K, et al. Long-term outcomes following intraosseous basivertebral nerve ablation for the treatment of chronic low back pain: 5-year treatment arm results from a prospective randomized double-blind sham-controlled multi-center study. Eur Spine J. 2020;29(8):1925-34. doi.org/10.1007/s00586-020-06448-x
Results from clinical studies are not predictive of results in other studies. Results in other studies may vary.
Warning: Stimulation modes. Only paresthesia-based stimulation mode has been evaluated for effectiveness in the diabetic peripheral neuropathy (DPN) population