
Cardiomiopatía
¿Qué es la Cardiomiopatía?
La cardiomiopatía afecta el miocardio en sí mismo. “Cardio” significa corazón y “miopatía” significa enfermedad del músculo. Existen varios tipos de cardiomiopatía. Las dos formas más comunes son la cardiomiopatía dilatada y la cardiomiopatía hipertrófica.
Las personas con cardiomiopatía a menudo presentan cardiopatías más graves:
- La cardiomiopatía dilatada (CMD) con frecuencia puede convertirse en una insuficiencia cardíaca.
- La cardiomiopatía hipertrófica (CMH) puede conducir a ritmos cardíacos acelerados e incluso a la muerte repentina de causa cardíaca.
- La displasia ventricular derecha arritmogénica (ARVD), un tipo poco frecuente de cardiomiopatía en la que el miocardio se reemplaza con grasa y reduce su capacidad para bombear sangre.
Reciba tratamiento, si lo necesita
Tanto la cardiomiopatía dilatada como la cardiomiopatía hipertrófica pueden conducir a cardiopatías más graves, como la insuficiencia cardíaca y muerte repentina de causa cardíaca.
Su médico puede decirle si padece CMD o si se encuentra en riesgo de presentar CMH. Saber cuáles son sus riesgos es el primer paso hacia el tratamiento. Además, el tratamiento adecuado a menudo puede ayudarlo a vivir más y mejor.
Causas
¿Qué causa la cardiomiopatía dilatada (CMD)?
¿Qué causa la cardiomiopatía hipertrófica (CMH)?
¿Qué causa la displasia ventricular derecha arritmogénica (ARVD)?
Se han identificado mutaciones en siete genes en el 40% o 50% de los individuos con ARVD3. (Estos genes incluyen DSC2, DSG2, DSP, JUP, PKP2, RYR2 y TMEM43.) La ARVD se hereda de manera autosómica dominante. Esto significa que si usted tiene ARVD, existe un 50% de probabilidad de transmitir el trastorno genético a cada uno de sus hijos.
Síntomas
Síntomas de la cardiomiopatía dilatada (CMD)
Síntomas de la cardiomiopatía hipertrófica (CMH)
Los síntomas de la CMH varían, pero pueden incluir dolor en el pecho, mareos o desvanecimiento.
Personas de cualquier edad pueden presentar cardiomiopatía hipertrófica. Para las personas jóvenes, desafortunadamente, la CMH a menudo causa muerte súbita cardíaca, incluso antes de saber que tienen CMH. De hecho, la miocardiopatía hipertrófica es la causa más común de muerte súbita cardíaca en personas menores de 30 años.
Si usted tiene un familiar con CMH, la muerte súbita cardíaca puede ser un riesgo grave. Hable con su médico acerca de la prueba.3
Síntomas de la displasia ventricular derecha arritmogénica (ARVD)
- Desmayos (síncope).
- Mareos.
- Latidos irregulares o acelerados (arritmia).
- Paro cardíaco repentino.
Si presenta síntomas de ARVD, éstos generalmente aparecen entre los 20 y los 40 años de edad, aunque se han observado síntomas en todas las edades. La ARVD por lo general se diagnostica mediante exámenes como el ecocardiograma para examinar la estructura del ventrículo derecho y evaluar cómo está funcionando.
Usted conoce su cuerpo lo suficientemente bien para saber cuándo siente algo extraño. Dado que los síntomas de la ARVD son similares a los de otras cardiopatías, debe consultar a su médico si experimenta alguno de estos síntomas.
Diagnóstico
¿Qué es la Cardiomiopatía Dilatada?
La cardiomiopatía dilatada (CMD) es el tipo más frecuente de cardiomiopatía. En la CMD, el miocardio se agranda, como lo indica en su nombre la palabra "dilatada".
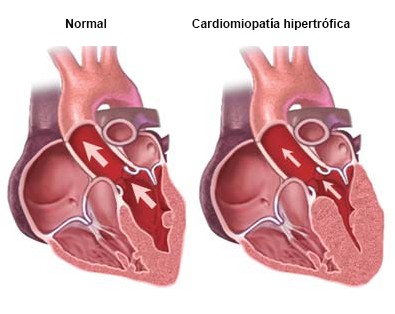
En la mayoría de los casos, la CMH se hereda de los padres o es el resultado de un problema con un gen antes del nacimiento. En cualquiera de las dos situaciones, las personas que padecen CMH pueden pasarle el gen a sus hijos. En otros casos, la CMH puede ocurrir a causa de la presión arterial alta o el envejecimiento.
Los síntomas de la CMH varían, pero pueden incluir dolor torácico, mareos o desvanecimiento.
Personas de todas las edades pueden presentar cardiomiopatía hipertrófica. En los más jóvenes, lamentablemente la CMH a menudo provoca muerte repentina de causa cardíaca, antes de que los pacientes sepan que la padecen. De hecho, la cardiopatía hipertrófica es la causa más común de muerte repentina de causa cardíaca en personas de menos de 30 años.1
Si un miembro de su familia tiene CMH, la muerte cardíaca repentina puede ser un riesgo grave. Hable con su médico acerca de realizarse exámenes.
¿Cómo se trata la CMH?
Con frecuencia, las personas con CMH tienen síntomas leves y pueden tener una esperanza de vida normal. Sin embargo, los pacientes generalmente reciben tratamientos si presentan síntomas o se encuentran en riesgo de sufrir una muerte repentina de causa cardíaca. Los tratamientos incluyen las siguientes opciones:
- Medicamentos: Su médico puede recetarle un betabloqueador que ayude a disminuir el endurecimiento del corazón.
- Cirugía: Algunos pacientes pueden someterse a una cirugía para extraer la parte endurecida del miocardio.
- Dispositivos cardíacos: Las personas en alto riesgo de sufrir una muerte repentina de causa cardíaca podrían recibir un desfibrilador implantable. El desfibrilador está previsto para detener una arritmia peligrosa antes de que cause muerte repentina de causa cardíaca.
¿Qué es la displasia ventricular derecha arritmogénica (ARVD)?
ARVD con CDI hasta trasplante
Chelsey Cornwall
La imagen anterior muestra las características histológicas típicas de la ARVC/D. Pérdida continua de miocitos (puntos negros) con fibrosis temprana e infiltración de adipocitos (áreas blancas rodeadas de miocardio rosado). (Utilizado con autorización. Gentileza de Thiene et al. Orphanet Journal of Rare Diseases 2007 2:45 doi:10.1186/1750-1172-2-45.)
Se calcula que la ARVD afecta a una entre 5000 personas. La enfermedad puede afectar tanto a hombres como a mujeres. Si bien es una causa relativamente poco frecuente de muerte repentina por causa cardíaca, representa hasta un quinto de las muertes repentinas por causas cardíacas en personas de menos de 35 años de edad. La ARVD también está asociada al paro cardíaco repentino en los atletas.
Los síntomas de la ARVD incluyen:
- Desmayos (síncope)
- Mareos
- Latidos acelerados o irregulares (arritmia)
- Muerte repentina por causa cardíaca
Si presenta síntomas de ARVD, éstos generalmente aparecen entre los 20 y los 40 años de edad, aunque se pueden observar síntomas en todas las edades. La ARVD por lo general se diagnostica mediante exámenes como el ecocardiograma para examinar la estructura del ventrículo derecho y evaluar cómo está funcionando.
Usted conoce su cuerpo lo suficientemente bien para saber cuándo siente algo extraño. Dado que los síntomas de la ARVD son similares a los de otras cardiopatías, debe consultar a su médico si experimenta alguno de estos síntomas.
¿Cómo se trata la ARVD?
Tratamiento
¿Cómo se trata la Cardiomiopatía Dilatada (CMD)?
Muchos de los tratamientos para la miocardiopatía dilatada son los mismos que los tratamientos para la insuficiencia cardíaca:
- Cambios en el estilo de vida: Su médico puede sugerirle que reduzca la cantidad de sal (sodio) en su dieta y que haga ejercicio todos los días.
- Medicamentos: Los beta bloqueadores y los inhibidores ECA (enzima convertidora de angiotensina) son dos medicamentos que usan con frecuencia para tratar la CMD.Si presenta edema, el médico también puede recetarle diuréticos.
- Dispositivos cardíacos : Si la cardiomiopatía hace que el corazón bombee de forma descoordinada, es posible que le coloquen un dispositivo implantable de terapia de resincronización cardíaca (TRC). Este dispositivo envía pequeñas cantidades de energía eléctrica al corazón que lo ayudan a volver a coordinar el bombeo. Si se encuentra en riesgo de padecer arritmias peligrosas, también podrían colocarle un cardiodesfibrilador implantable. El desfibrilador puede detener la arritmia antes de que provoque la muerte repentina de causa cardíaca.
- Cirugía: Es posible que algunos pacientes necesiten cirugía para reparar una válvula cardíaca. O bien, los médicos pueden considerar un trasplante de corazón en pacientes con síntomas muy intensos.
¿Cómo se trata la Cardiomiopatía Hipertrófica (CMH)?
A menudo las personas con CMH tienen síntomas suaves y muchos pueden esperar tener una vida útil normal. Sin embargo, los pacientes generalmente reciben tratamientos si presentan síntomas o se encuentran en riesgo de sufrir una muerte repentina de causa cardíaca. Los tratamientos incluyen las siguientes opciones:
- Medicamentos: Su médico puede recetarle un betabloqueador que ayude a disminuir el endurecimiento del corazón.
- Cirugía: Algunos pacientes pueden someterse a una cirugía para extraer la parte endurecida del miocardio.
- Dispositivos cardíacos: Las personas en alto riesgo de sufrir una muerte repentina de causa cardíaca podrían recibir un desfibrilador implantable. El desfibrilador está diseñado para detener una arritmia peligrosa antes de que cause una muerte súbita cardíaca.
Obtener tratamiento si es necesario
Tanto la cardiomiopatía dilatada como la cardiomiopatía hipertrófica pueden conducir a cardiopatías más graves, como la insuficiencia cardíaca y muerte repentina de causa cardíaca.
Su médico puede decirle si padece CMD o se encuentra en riesgo de presentar CMH. Saber cuáles son sus riesgos es el primer paso hacia el tratamiento. Además, el tratamiento adecuado a menudo puede ayudarlo a vivir más y mejor.